We work at the interface of chemistry and machine learning to develop models that understand how molecules behave, interact, and react and use that knowledge to engineer new ones. Much of our work focuses on improving computational strategies for small molecule drug discovery, molecular optimization, synthesis planning, and structure elucidation. A long-term goal of our work is to enable autonomous molecular discovery, where hypotheses are proposed algorithmically and tested via experiments with minimal human intervention.
Small molecules are the predominant modality for medicines, chemical probes, organocatalysts, and specialty monomers among others. They are typically discovered through an iterative process of designing candidate compounds, synthesizing them, and testing their performance, where each repeat of the cycle requires weeks or months. The rate at which this process yields successful compounds can be limited by bottlenecks and mispredictions at all three stages and is plagued by inefficiencies, including underutilization of available data resulting in inadequate predictions of compound performance, compound selection based on intuition or synthetic ease rather than information content, and frequent manual intervention subject to human bias. For example, the hit-to-lead and lead optimization stages of small molecule drug discovery, while only part of the overall pipeline, require several years and millions of dollars for each clinical candidate. Even longer timescales are required to bring a new material to market.
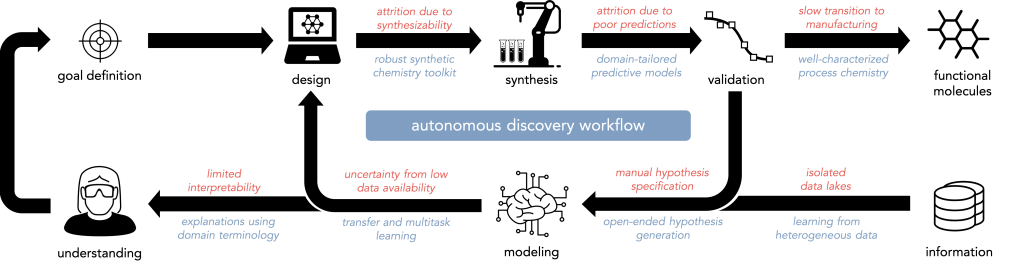
The discovery of these molecules—and scientific discovery more generally—is a problem of inference from incomplete and imperfect information, for which techniques in artificial intelligence are well-suited. However, there are a number of bottlenecks in our current approach to molecular discovery (a few are summarized below in red); overcoming them will require a number of methodological advances (a few are summarized below in blue).
For a slightly-outdated comprehensive overview of autonomous discovery in the chemical sciences, including a discussion of key challenges, please read our 2019 review article and perspective in Angewandte Chemie or on arxiv: part one, part two. Our work falls under the broader umbrella of “AI for Science”.
One of the most important questions one must answer when searching for a molecule that achieves a certain property profile is ‘what should we make next?’ out of the vastness of chemical space. Molecules are generally proposed through one of two complementary approaches: virtual screening, where one has a fixed list of candidates, and generative modeling, where one uses algorithms to propose novel molecular structures. Exhaustive virtual screening with a computational oracle (e.g., docking for structure-based drug design) is a viable approach to hit finding, but is not straightforward to apply to the tens of billions of molecules in modern virtual libraries. We work on various methods for selecting and proposing molecular structures during iterative rounds of optimization, both for from discrete libraries and using generative models. The former uses techniques in model-guided optimization (Bayesian optimization) to navigate large discrete design spaces of candidate molecular structures. A thread of particular interest is the development of synthesizability-constrained generative models that propose molecular structures that are synthetically accessible and/or using information about synthesizability as the basis for the downselection of candidates. Sample efficiency is also a primary concern for these moodels, as in silico benchmarks may involve the ‘testing’ of hundreds of thousands of molecules, which is severely misaligned with what is practical, experimentally. Finally, we have sought to better integrate principles of medicinal chemistry and noncovalent interactions into generative design to overcome yet another failure mode of generative models, which is the relatively poor quality of ligands they generate in 3D pocket-conditioned generative tasks.
Selected publications (screening/selection/non-generative):
Selected publications (generative design):
Computational assistance is only useful insofar as it can produce actionable suggestions for experimental validation. The synthesis and testing of new molecules is a common requirement for validation of functional physical matter. Despite significant progress in methodology development, few reactions have achieved widespread use as part of the synthetic chemistry toolbox. Newly discovered reactions may be too narrowly applicable or require excessive optimization to be practical. Overcoming both of these barriers to adoption requires an expansion of substrate scope and straightforward guidelines for how the reaction should be carried out to achieve maximal yield, selectivity, etc. We approach this problem from a statistical modelling perspective, leveraging the collective chemical knowledge reflected in the patent and journal literature to identify robust synthetic and biological transformations, including enzyme catalysis. This builds on our ongoing efforts in ‘predictive chemistry’, which encompasses computer-aided synthesis planning, reaction outcome prediction, and related tasks. Our work in this area also explores the intersection of structure-based representation learning and descriptor-based approaches that have become prevalent in physical organic chemistry. A particular challenge is transforming these methods from being qualitative to quantitative, and retrospective to prospective. To support our more ambitious goals of extrapolation, we also develop models that are designed to predict mechanistic pathways with the expectation that a grounding in mechanistic understand may help models generalize to previously-unseen reaction types.
Several data-driven chemistry tools for synthesis planning are available in the open source ASKCOS software suite, which has seen use by 35,000+ chemists and is deployed at 15+ pharmaceutical and chemical companies. Try out the public deployment at askcos.mit.edu or spin up your own deployment using these directions.
Selected publications (retrosynthesis-focused):
Selected publications (outcome prediction-focused):
Small molecule metabolites mediate myriads of biological and environmental phenomena across host-microbiome interactions, plant chemistry, cancer biology, and various other processes. Mass spectrometry is often used as an analytical technique to investigate the small molecules present in a sample, measuring both their masses and fragmentation spectra. However, the complexity and high dimensionality of spectral data makes it difficult to identify unknown metabolites and their roles, with a large majority of detected metabolites remaining unidentified in public data. We are developing a suite of new computational methodologies for higher accuracy annotation of small molecule metabolites from mass spectrometry data that integrate chemistry-informed priors with modern deep learning advancements. These include a variety of forward models as well as inverse models that try to solve the structure-to-spectrum and spectrum-to-structure tasks, respectively. The latter includes methods that treat elucidation as a conditional molecular design task that is therefore amenable to many of the optimization-based and generative approaches we develop in the context of small molecule drug design as well.
Selected publications:
The use of machine learning models for chemical discovery problems has overwhelmingly been limited to off-the-shelf models that do not readily transfer to this domain. Problems in chemistry and biology rarely have large quantities of clean, annotated data, and the structure of model inputs (e.g., molecules, proteins) are not inherently numeric or character-based as in more familiar image-or text-processing tasks. While statistical inference techniques have been successfully applied to self-contained chemical prediction tasks (e.g., developing structure-property relationship models, predicting links in knowledge graphs of biological activity), there are significant obstacles to applying them more broadly related to molecular representation, model generalizability, and data availability, among others. Our work in this area involves the integration of domain expertise and prior knowledge into neural models to improve accuracy, uncertainty estimation, and generalization, especially in low data environments. >2D molecular representations that can understand both stereochemistry and conformational flexibility are of particular interest. Beyond small molecules, we also think about representation learning for molecular ensembles (e.g., polymers) and porous materials (e.g., zeolites).
Selected publications:
As computational design strategies are increasingly able to make use of available information to propose new hypotheses, we are increasingly limited by our ability to validate them. More bluntly, synthesis is the bottleneck for many molecular discovery workflows. Current platform technologies for synthesis and screening are often misaligned with rational design approaches; the next generation of laboratory hardware should be specifically tailored to supply the data required by the current and future state-of-the-art machine learning and computational discovery algorithms. We customize our own automated laboratory platforms for the generation of high-fidelity experimental data suitable for training and validation. Our intent is to integrate automated laboratory hardware with our computational models to autonomously conduct reaction and molecule screening campaigns, inching closer to a true ‘closed-loop’ vision beyond the proofs of concept the field has seen to date. Even if we are far from true autonomy, automated platforms are able to explore larger design spaces with better time- and material-efficiency than traditional manual experimentation.
Selected publications:
The development of data-driven methods relies on data. At present, most procedural details about chemical reactions are reported in unstructured supporting information documents as Word files exported to PDFs. Information is not fully captured by current databasing efforts including, even basic details of quantitative amounts. The state of data sharing for other molecular property prediction tasks is better, but efforts to standardize data splits, evaluations, and pre-processing pipelines are essential. In addition to our commitment to developing open source software for the research threads listed above, we help lead the Open Reaction Database and contribute to the Therapeutics Data Commons.
Selected publications: